单细胞上游分析
单细胞测序介绍
单细胞测序技术
通过在单个细胞水平对转录组或基因组进行扩增并测序,可以研究单个细胞内的基因表达情况,同时解决用组织样本测序无法解决的细胞异质性(如在肿瘤组织中,肿块中心的细胞,肿块周围的细胞,淋巴转移灶的细胞,以及远端转移的细胞,其基因组和转录组等遗传信息,是存在差异的。而这种差异,在临床上,可以决定该肿瘤对某种疗法是否有效)难题,让解析单个细胞的行为、机制及其与机体的关系成为了现实。
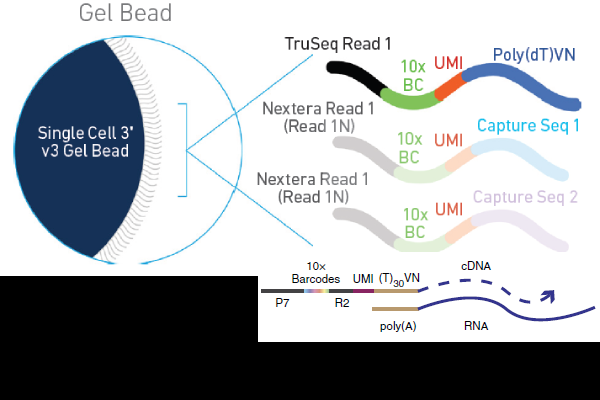
测序平台 10 X Genomics单细胞转录组测序平台
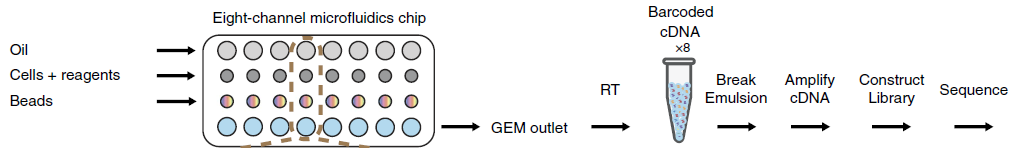
利用微流控、油滴包裹和Barcode标记等技术来实现高通量的细胞捕获技术,能够一次性分离、并标记500–10000个单细胞,从而获得每个细胞的3’端的转录组信息。具有细胞通量高、建库成本低、捕获周期短等优势。该技术主要用于细胞分型和标记因子的鉴定,从而实现对细胞群体的划分与细胞群体间基因表达差异的检测,此外该技术还可以预测细胞分化与研究发育轨迹,在当下疾病、免疫、肿瘤领域以及组织、器官、发育研究中发挥越来越重要的作用。

数据下载
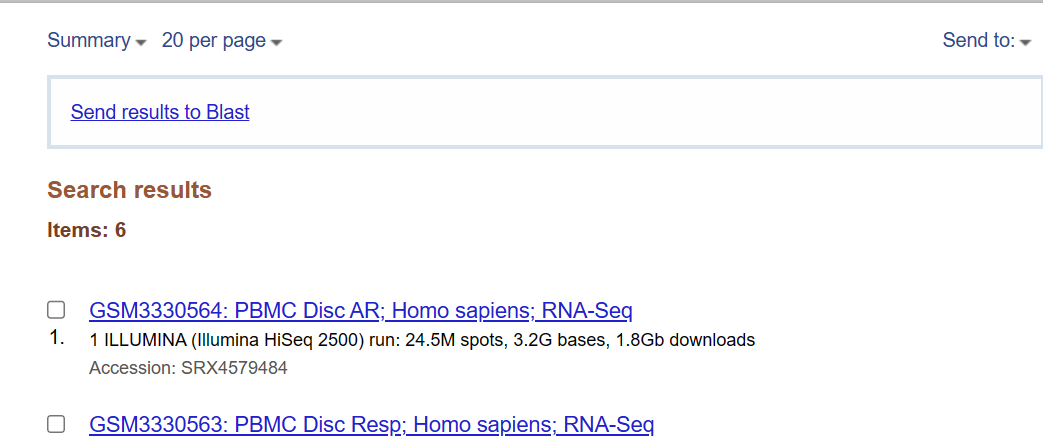
前往GEO数据库,得到目标样本的SRR ID号,本文以GSE117988为例,进行后续分析。
1、进入GEO数据库,找到relation中的SRA

2、下载为SRR_list文件
 send to ==> run selector ==> go
进入如下界面
send to ==> run selector ==> go
进入如下界面
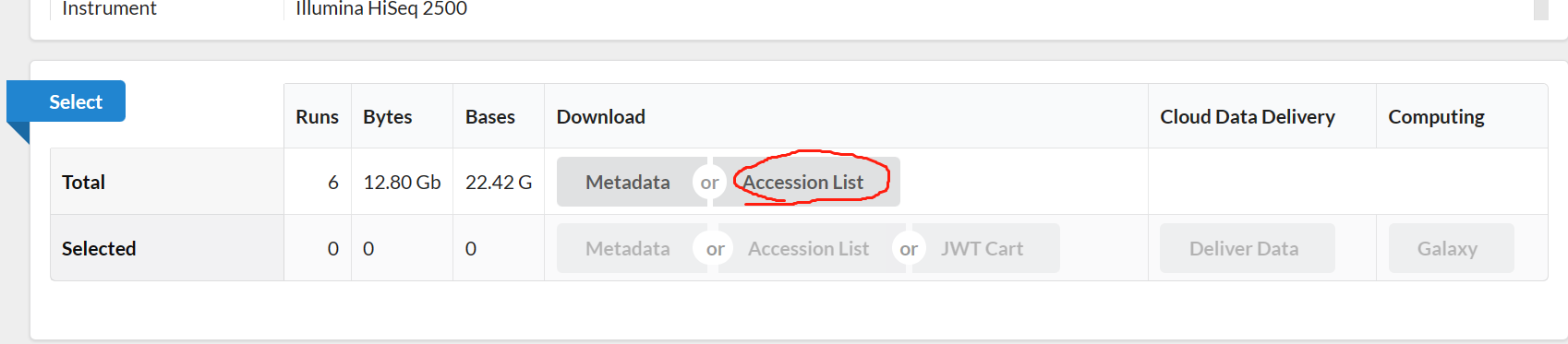 点击Accession List 进行下载,后上传List文件到服务其中
点击Accession List 进行下载,后上传List文件到服务其中
服务器相关软件设置
1、sratoolkit和aspera下载
下载数据时我们一般使用sratools中的prefetch,但是直接使用prefetch会速度非常的慢。当我们配上aspera时,下载速度就会变快。
##下载sraroolkit
cd ~/biosoft
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.2/sratoolkit.3.0.2-ubuntu64.tar.gz
tar -zxvf sratoolkit.3.0.2-ubuntu64.tar.gz
echo 'export PATH=/home/data/t070401/biosoft/sratoolkit.3.0.2-ubuntu64/bin:$PATH'>> ~/.bashrc # 写入环境变量中
source ~/.bashrc ## 保存一下
## 查看安装成功没
prefetch -h # 弹出使用说明就是成功了
# 下载ascp
cd ~/biosoft
wget http://download.asperasoft.com/download/sw/connect/3.7.4/aspera-connect-3.7.4.147727-linux-64.tar.gz
tar zxvf aspera-connect-3.7.4.147727-linux-64.tar.gz
#安装
bash aspera-connect-3.7.4.147727-linux-64.sh
# 然后cd到根目录下看看是不是存在了.aspera文件夹,有的话表示安装成功
cd && ls -a
# 将aspera软件加入环境变量,并激活
echo 'export PATH=~/.aspera/connect/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
# 最后检查ascp是不是能用了
ascp --help
2、安装cellranger
cellranger的下载链接具有时效性,可以去10X官网下载
(https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest)
cd ~/biosoft
wget -O cellranger-7.1.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-7.1.0.tar.gz?Expires=1681409686&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1leHAvY2VsbHJhbmdlci03LjEuMC50YXIuZ3oiLCJDb25kaXRpb24iOnsiRGF0ZUxlc3NUaGFuIjp7IkFXUzpFcG9jaFRpbWUiOjE2ODE0MDk2ODZ9fX1dfQ__&Signature=g06lNWAxunjOQbd~LKht32n4RG9K0UpWMGhwF7mVznbn45BAjhA5aNONcNMxIHLh8Rwelgf1LxnLxoFQF4aQb7j5EPKwcTbrgaX8ndlA0vR7zXVkiXroQ0NmEREeyni75QrNbw-~e5UoEFo9EjaKWOQuGOjL4drjDUQoVWJNmi4ONFQkvX6cs-8QJ4uVIaVecVjjWD6JkQWtyGyFiGXHIYUh0aWFrpFkZzh4IURDS8C6dkaBPZV5rYFujuRSBsFtS3NQKF8aiBMqUVk5T43wScm60KN5CNBhfZGsz3K92l3F9~-FnGnmUhwBJHlC4aO2C79k7a6tX0LnM038jAfn4w__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
# 校对一下md5码是否一致
md5sum cellranger-7.1.0.tar.gz
# 解压
tar -zxvf cellranger-7.1.0.tar.gz
echo 'export PATH=/home/data/t070401/biosoft/cellranger-7.1.0/bin:$PATH'>> ~/.bashrc # 写入环境变量中
source ~/.bashrc ## 保存一下
## 查看安装成功没
cellranger -h # 弹出使用说明就是成功了
3、创建专属conda环境
#conda install mamba
## 使用mamba下载包会非常的快
conda create -n 10x
conda activate 10x
mamba install -y -c bioconda aspera-cli bwa samtools bedtools sambamba sra-tools bowtie2 samblaster fasterq-dump
4、下载参考基因组
cd ~/single_cell/10x_refdata
## mm10
nohup wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz & # 挂nohup & 让他在后台下载
md5sum refdata-gex-mm10-2020-A.tar.gz
#886eeddde8731ffb58552d0bb81f533d refdata-gex-mm10-2020-A.tar.gz
tar -xzvf refdata-gex-mm10-2020-A.tar.gz
####################################################################################################################################################
## hg38
nohup wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz &
md5sum refdata-gex-GRCh38-2020-A.tar.gz
#dfd654de39bff23917471e7fcc7a00cd refdata-gex-GRCh38-2020-A.tar.gz
tar -xzvf refdata-gex-GRCh38-2020-A.tar.gz
分析流程
sra数据下载
创建分析所用的文件夹
cd ~/single_cell/cellranger
mkdir 1.sra 2.raw_fastq 3.cellranger
把下载好的SRR list放入到1.sra文件夹下
cd ./1.sra
cat SRR_Ac_List.txt |while read id;do (nohup prefetch $id &);done
1、SRA 转 fastq
常规的SRA转fastq文件,用的是fastq-dump软件,由于该软件是使用单线程,速度非常慢。fasterq-dump可以加快速度,但是可能时版本不匹配,无法正常使用。为此,我们使用parallel-fastq-dump进行数据的下载
conda activate 10x
pip install parallel-fastq-dump
Linux ln(英文全拼:link files)命令是一个非常重要命令,它的功能是为某一个文件在另外一个位置建立一个同步的链接。
当我们需要在不同的目录,用到相同的文件时,我们不需要在每一个需要的目录下都放一个必须相同的文件,我们只要在某个固定的目录,放上该文件,然后在 其它的目录下用ln命令链接(link)它就可以,不必重复的占用磁盘空间。 这里为我们为1.sra文件夹中的数据建立一个到2.fastq的链接
cd 2.raw_fastq
ln -s ../1.sra/SRR* ./
#写一个脚本批量进行转换
vim fastq.sh
cat ../1.sra/SRR_Acc_List.txt | while read id;do (nohup parallel-fastq-dump --gzip --threads 4 --outdir ./ --split-files -s ./$id/$id.sra& );done
--split-files 该参数可以将sra文件拆分为3个文件
--gzip 对结果进行压缩
--threads 使用多少个线程 这里设置为4
此外这里的**--split-files**若换成**--split-3**
如果结果发现只有一个文件,说明数据不是双端(第三个文件太大会覆盖前两个);
如果结果有两个文件,说明是双端文件并且数据质量比较高(没有低质量的reads或者长度小于20bp的reads);
如果结果有三个文件,说明是双端文件,但是有的数据质量不高,存在trim的结果,第三个文件的名字一般是:<srr_id>.fastq, 而且文件也不大,基本可以忽略。
在cellranger流程中,为了后续分析,我们采用**--split-files**。
bash fastq.sh ##!!! 勿忘ps查看一下进程
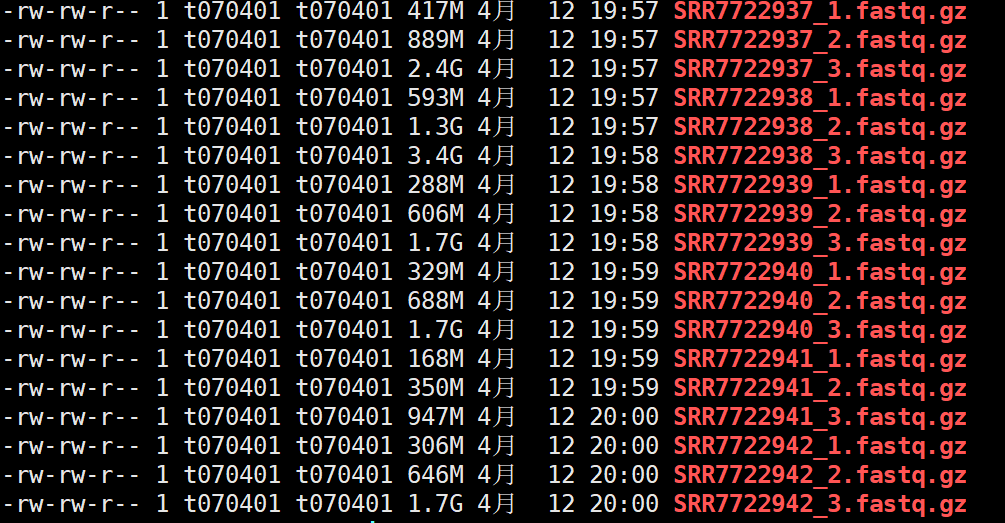
Run cellranger
先把文件改一下名
vim change_name.sh
cat ../1.sra/SRR_Acc_List.txt|while read i;do (mv ${i}_1*.gz ${i}_S1_L001_I1_001.fastq.gz;
mv ${i}_2*.gz ${i}_S1_L001_R1_001.fastq.gz;
mv ${i}_3*.gz ${i}_S1_L001_R2_001.fastq.gz);done
bash change_name.sh
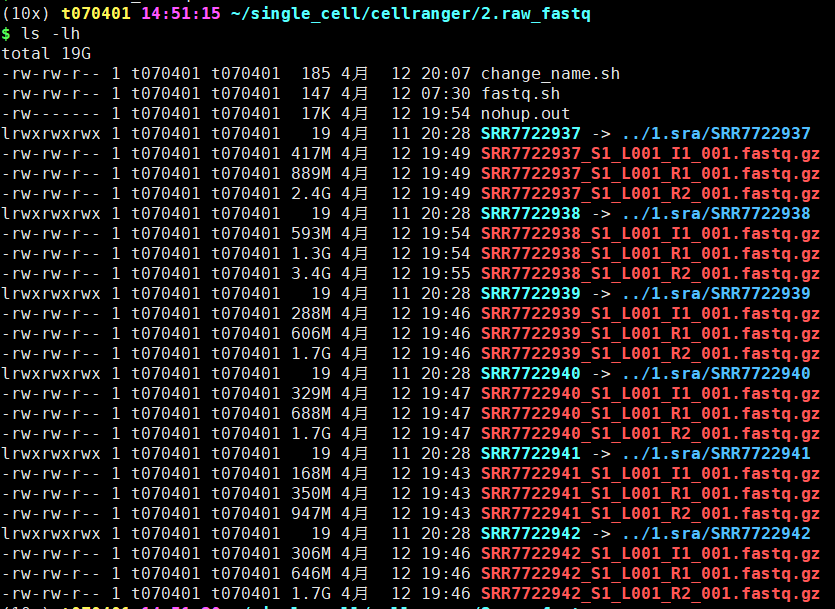
然后就是走cellranger啦
cd ../3.cellranger
# 同理也可以写一个shell脚本
# 我们的样本是人的,就用这个ch38的参考基因组
vim run_cellranger.sh
ref=/home/data/t070401/single_cell/10x_refdata/refdata-gex-GRCh38-2020-A
cat ../1.sra/SRR_Acc_List.txt|while read id;
do (nohup cellranger count --id=$id \
--transcriptome=$ref \
--fastqs=/home/data/t070401/single_cell/cellranger/2.raw_fastq \
--sample=$id \
--nosecondary \
--localcores=4\
--localmem=30 &);
done
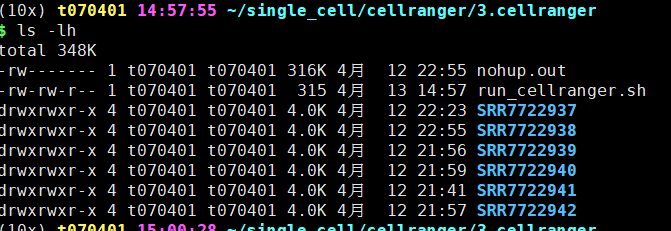
bash run_cellranger.sh
## !! 勿忘ps一下进程
最主要的几个参数:
--id文件夹的名字
--transcriptome 指定参考基因组的路径
--sample 指定需要处理的fastq文件的前缀
--expect-cell 指定预期的细胞数目,默认参数是3000个
--localcores 指定计算的核心数
--mempercore 指定内存大小 GB
--nosecondary 不需要进行降维聚类(因为后期会用R可视化)
--chemistry,默认是自动识别chemistry,但是有些时候识别不出chemistry的时候,需要加入参数特别标明
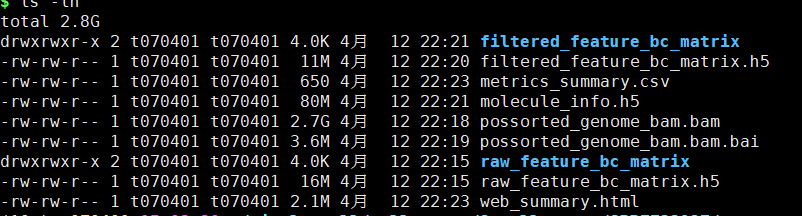
最后得到结果在outs文件夹下
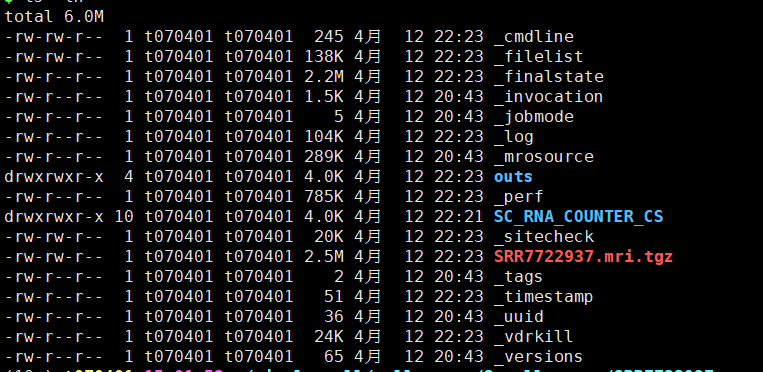
 得到的结果就可以用seurat读取然后进行下游分析啦。
得到的结果就可以用seurat读取然后进行下游分析啦。
结果解读
web_summary.html:必看,官方说明 summary HTML file ,包括许多QC指标,预估细胞数,比对率等;
metrics_summary.csv:CSV格式数据摘要,可以不看;
possorted_genome_bam.bam:比对文件,用于可视化比对的reads和重新创建FASTQ文件,可以不看;
possorted_genome_bam.bam.bai:索引文件;
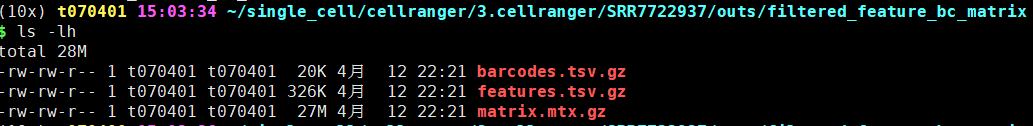
filtered_gene_bc_matrices:是最为重要的一个目录,下面又包含了 barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,是下游Seurat、Scater、Monocle等分析的输入文件,是经过Cell Ranger过滤后构建矩阵所需要的所有文件;
filtered_feature_bc_matrix.h5:过滤掉的barcode信息HDF5 format,可以不看;
raw_feature_bc_matrix:原始barcode信息,未过滤的可以用于构建矩阵的文件,可以不看;
raw_feature_bc_matrix.h5:原始barcode信息HDF5 format,可以不看;
analysis:数据分析目录,下面又包含聚类clustering(有graph-based & k-means)、差异分析diffexp、主成分线性降维分析pca、非线性降维tsne,因为我们自己会走Seurat流程,所以不用看;
molecule_info.h5:可用于整合多样本,使用cellranger aggr函数;
cloupe.cloupe:官方可视化工具Loupe Cell Browser 输入文件,无代码分析的情况下使用,会代码的同学通常用不到。